
L’hémoglobine A est formée de la globine et de l’hème et est capable de capter l’oxygène au niveau pulmonaire et de le relarguer aux tissus. Elle est constituée de 4 chaînes (2 chaînes α et 2 chaînes β et de 4 groupements hème.
Clinique de la drépanocytose
Sommaire
Qu'est-ce-que la drépanocytose?
Quelles sont les différentes formes de syndromes drépanocytaires majeurs?
Qu'est-ce que l'hémoglobine A?
Quelle est l'anomalie génétique de la drépanocytose?
Comment l'hémoglobine S intervient-elle?
Quels sont les facteurs génétiques modifiant la symptomatologie?
Quels sont les signes cliniques chez l'enfant?
Références
Mise à jour 28 janvier 2018
QU’EST-CE QUE LA DREPANOCYTOSE ?
Il s’agit de la maladie génétique la plus fréquente dans le monde. Le dépistage néonatal ciblé sur les populations à risque et généralisé à toute la France depuis 2000 a montré qu’environ 330 nouveaux-nés étaient atteints de syndrome drépanocytaire majeur en France ce qui en fait la plus fréquente des maladies génétiques dépistées à la naissance. Elle concerne principalement les enfants ayant une ascendance africaine. Il s’agit d’une hémoglobinopathie dans laquelle l’Hémoglobine A normale est remplacée par l’Hémoglobine S polymérisant en situation d’hypoxie et aboutissant à une rigidification du globule rouge. Cette maladie se caractérise par trois sortes de complications principales : une hémolyse responsable de l’anémie et de l’ictère, un risque thrombotique exposant aux accidents ischémiques, osseux et de tous les organes (poumons, rate, reins, cerveau, cœur etc.) et un risque infectieux lié à l’asplénie fonctionnelle.
La période de l’enfance est dominée par les risques infectieux, le risque de séquestration splénique, la survenue de crises douloureuses vaso-occlusives et celui de vasculopathie cérébrale tandis que chez l’adulte surviennent les problèmes de défaillance organique. Le dépistage néonatal permet la mise en route précoce de la prophylaxie anti-pneumococcique et l’éducation parentale permettant d’éviter bon nombre de complications. Des progrès majeurs ont été réalisés grâce à la détection précoce de la vasculopathie cérébrale par le Doppler transcrânien et la mise en route de thérapeutiques adaptées. L’hydroxyurée, les programmes transfusionnels et l’allogreffe géno-identique de cellules souches hématopoïétiques ont considérablement amélioré la qualité de vie. Par ailleurs, un protocole de thérapie génique va très prochainement être initié. La qualité de la prise en charge dans l’enfance favorise une bonne insertion socioprofessionnelle à l’âge adulte.
QUELLES SONT LES DIFFERENTES FORMES DE SYNDROMES DREPANOCYTAIRES MAJEURS ?
Les syndromes drépanocytaires majeurs regroupent 3 types d’hémoglobinopathie : les formes homozygotes SS, les doubles hétérozygoties SC et Sβthalassémie et d’autres formes plus rares telles que la SDPunjab
QU’EST-CE QUE L’HEMOGLOBINE A ?
|
L’hémoglobine A est formée de la globine et de l’hème et est capable de capter l’oxygène au niveau pulmonaire et de le relarguer aux tissus. Elle est constituée de 4 chaînes (2 chaînes α et 2 chaînes β et de 4 groupements hème. |
QUELLE EST L’ANOMALIE GENETIQUE DE LA DREPANOCYTOSE ?
La drépanocytose est une maladie génétique autosomale récessive dans laquelle l’hémoglobine A normale α2 β2 est remplacée par l’hémoglobine α2 β2Σ produit d’une mutation génique sur le gène de la globine β substituant au niveau du 6ème codon une adénine par une thymidine, aboutissant au remplacement d’un acide glutamique en valine β6glu\val).
|
Dans sa forme homozygote SS, les deux parents sont généralement tous deux porteurs AS, asymptomatiques et ont un risque de 25% d’engendrer un enfant SS à chaque conception. Les patients SS ne fabriquent pas d’HbA1 et seulement de l’HbF α2 γ2 en quantité variable et de l’HbA2. L’union d’un Parent AS avec un parent porteur d’une β0thalassémie mineure (hétérozygote) risque également d’engendrer dans 25% des cas un enfant atteint de thalasso-drépanocytose Sβ0-thalassémie), ne fabriquant pas non plus d’HbA1 remplacée par de l’HbS et dont la symptomatologie est équivalente à celle d’une forme SS.
|
On classe également dans les syndromes drépanocytaires majeurs les formes SC provenant de l’union d’un parent AS et d’un autre AC (risque de 25% d’enfant SC) et les formes S/β+thalassémie provenant de l’union d’un parent AS avec un porteur de β+thalassémie mineure. Il se trouve que les porteurs AS ont une résistance accrue aux formes de neuropaludisme ce qui a contribué au maintien et même à l’avantage sélectif de cette mutation dans les pays impaludés. Ainsi, 300000 nouveaux-nés atteints de drépanocytose SS naissent chaque année dans le monde, en particulier dans les populations africaines, dans certaines régions méditerranéennes, au Moyen-Orient et en Asie (Inde) ainsi qu’aux Antilles et Amériques.
COMMENT L’HEMOGLOBINE S INTERVIENT-ELLE ?
L’ hémoglobine S a comme caractéristique majeure de polymériser en situation désoxygénée conduisant à la formation de fibres rigides déformant (en faucille d’où le nom de falciformation donné à cette déformation), rigidifiant et lésant le globule rouge drépanocytaire, diminuant ainsi sa durée de vie. En dehors de l’hypoxie, les autres facteurs favorisant la polymérisation de l’HbS sont la déshydratation et l’acidose. Dans un premier temps, la situation est réversible et l’HbS peut être resolubilisée par apport d’oxygène, hydratation et alcalinisation qui seront largement utilisés sur le plan thérapeutique.
La symptomatologie sera liée à l’association : hémolyse-anémie (ictère, asthénie, hypotrophie, souffrance tissulaire par hypoxie chronique..) et aux épisodes aigus de vaso-occlusion générateurs de crises douloureuses. Les drépanocytes générés par les conditions d’hypoxie, adhèrent anormalement à l’endothélium, en particulier dans les veinules, les granulocytes et les macrophages y sont activés ce qui engendre une obstruction aiguë de la microcirculation avec réaction inflammatoire . Les phénomènes vaso-occlusifs peuvent toucher tous les organes (infarctus osseux, pulmonaires, priapisme, rétinopathie, AVC….) mais ils surviennent tout particulièrement au niveau splénique et sont responsables de l’asplénie fonctionnelle observée dès les premiers mois de vie dans les formes SS. Cette asplénie fonctionnelle est à l’origine du risque infectieux majeur dans cette pathologie : risque de septicémies foudroyantes en particulier à pneumocoques mais aussi à Salmonelles, Hémophilus, et plus rarement à bacilles gram négatifs et risque accru d’infections à Mycoplasmes. Les complications infectieuses peuvent être associées aux infarctus et leur diagnostic est souvent difficile du fait de la réaction inflammatoire présente au cours de toute crise. Seuls les prélèvements bactériologiques permettront d’apporter la preuve de la surinfection.
QUELS SONT LES FACTEURS GENETIQUES MODIFIANT LA SYMPTOMATOLOGIE ?
|
Chez l’hétérozygote AS, à l’intérieur du globule rouge, la présence d’HbA à côté de l’HbS inhibe la polymérisation de celle-ci et ces patients sont tout à fait asymptomatiques dans les conditions standard de vie et ont une espérance de vie normale. Seules des conditions extrêmes d’exercice et d’hypoxie sont susceptibles de déclencher des crises. Les patients SC et S/β+thalassémiques sont moins anémiques que les patients SS et n’ont pas de risque de vasculopathie cérébrale des gros vaisseaux, mais ont un risque accru de rétinopathie et d’ostéonécrose.
|
L’association à une α-thalassémie (délétion d’un ou deux gènes α) modifie le volume du globule rouge (microcytose) et diminue l’intensité de l’hémolyse et de l’anémie et le risque d’AVC.
L’étude du polymorphisme génétique de la chaîne β permet de distinguer les formes « Bantou » des formes « Sénégal » et « Bénin » : les formes « Bantou » sont les plus anémiques et en général plus sévères.
QUELS SONT LES SIGNES CLINIQUES CHEZ L’ENFANT ?
La clinique va être dominée chez l’enfant par les 3 aspects : hémolyse, anémie ; accidents vaso-occlusifs ; infections.
1. Chez le nourrisson
Le risque de manifestations précoces avant l’âge d’un an ne concerne pratiquement que les patients SS et est un témoin de sévérité de la maladie.
Avoir à l’esprit les deux grands dangers chez le nourrisson:
- le risque de pneumococcie fulminante (asplénie fonctionnelle)
- le risque de séquestration splénique
Le risque de séquestration splénique existe dès les premiers mois (il en a même été observé dès l’âge de 1 mois) : il s’agit d’une complication redoutable pouvant être fatale. Les drépanocytes sont trappés dans la rate qui grossit brutalement, entraînant ainsi une anémie majeure pouvant entraîner la défaillance cardiaque. Les signes cliniques d’appel sont la pâleur, l’anorexie, l’asthénie et il est capital d’avoir éduqué préalablement les parents à la palpation de la rate et de leur avoir demandé de venir à l’hôpital rapidement pour un contrôle sanguin en cas de doute. Seule, la transfusion simple va permettre de reverser ce phénomène en remettant en circulation les drépanocytes séquestrés (la splénomégalie régresse très rapidement au décours de la transfusion). Le risque de récidive est majeur et peut faire discuter la mise en route d’un programme transfusionnel jusqu’à l’âge de la splénectomie. L’HU s’est révélé être inefficace pour la prévention de ce risque qui existe même lorsque le taux d’HbF est encore élevé.
Le risque infectieux existe également très précocement, l’asplénie fonctionnelle pouvant survenir dès les premiers mois de vie
Il s’agit surtout d’un risque de septicémie foudroyante associée ou non à une atteinte méningée à pneumocoque. Ce risque justifie d’une part l’ensemble des mesures préventives prises consistant à introduire dès l’âge de 2 mois la prophylaxie par Pénicilline orale, et la vaccination par le Prévenar désormais disponible. Ce vaccin conjugué, dirigé contre les 7 valences les plus pathogènes, est en effet immunogène dès les premiers mois de vie, à la différence du Pneumo 23 qui n’est réellement efficace qu’à partir de l’âge de 2 ans (mais protège contre 23 valences). D’autre part, ce risque de septicémies foudroyantes impose de faire venir systématiquement à l’hôpital tous les nourrissons fébriles (> 38°5) pour y recevoir un bilan sanguin avec hémoculture et une antibiothérapie IV (il convient de couvrir contre les pneumocoques et les salmonelles et la Ceftriaxone est le souvent choisie. L’expérience a montré qu’il était insuffisant et imprudent de se fier à la clinique et même aux signes biologiques et que seule une attitude systématique permettait d’éviter les drames.
Toutes fièvres > 38°5 doivent à priori
faire craindre une pneumococcie
et faire décider l’hospitalisation pour antibiothérapie IV
Les premières crises vaso-occlusives surviennent le plus souvent sous forme du Syndrome Pieds-Mains
Il s’agit d’une crise douloureuse des extrémités avec un gonflement du dos des mains et des pieds associé souvent à un gonflement des doigts (dactylite) : plus ces crises surviennent tôt plus elles sont un signe de sévérité. Elles ne nécessitent pas forcément une hospitalisation en absence de fièvre importante et le plus souvent les mesures d’hydratation orale, et les antalgiques associant paracétamol, ibuprofène et codéine suffisent dans un premier temps. Par contre, la survenue secondaire d’une fièvre ou l’apparition de symptômes pulmonaires impose l’hospitalisation, toute crise vaso-occlusive étant susceptible de se compliquer secondairement du syndrome thoracique tant redouté.
2. Chez l’enfant
Les crises douloureuses vaso-occlusives dominent le tableau : souvent déclenchées par un effort physique trop intense, une séance de piscine, elles peuvent aussi survenir spontanément ou à l’occasion d’une obstruction naso-pharyngée. Elles concernent le plus souvent les membres et les articulations mais elles peuvent être aussi abdominales ou thoraciques touchant les côtes et le sternum. La douleur peut être isolée ou associée à une tuméfaction, érythémateuse avec augmentation de chaleur locale ne traduisant pas forcément une surinfection. Pour les crises douloureuses des membres, isolées et sans tuméfaction, le traitement est initié à la maison par l’apport de boissons abondantes (1/4 vichy ou coca, ¾ eau, paracétamol, nureflex, voire codéine) mais l’hospitalisation est nécessaire pour les crises trop intenses (nécessité de recourir à une hydratation IV et à la morphine), celles avec tuméfaction (de façon à éliminer une ostéomyélite), celles durant depuis plus de 48h ou s’accompagnant de fièvre > 38°5 ou surtout de symptômes thoraciques (douleur thoracique, toux, dyspnée, polypnée). Les deux complications peuvent être la surinfection (ostéomyélite ….en fait rare) mais surtout :
Le syndrome thoracique aigu défini par l’apparition d’un infiltrat radiologique pulmonaire associé à un ou plusieurs des signes suivants : toux, polypnée, dyspnée, douleur thoracique, hypoxie, fièvre. Il correspond à un phénomène vaso-occlusif aboutissant à un infarctus pulmonaire. L’occlusion vasculaire peut être due à une embolie graisseuse à partir du lieu de la CVO osseuse, à une thrombose favorisée par l’hypoventilation (elle-même en rapport avec la douleur thoracique ou au contraire avec la sédation morphinique). Par ailleurs les CVO s’accompagnent très fréquemment de déglobulisation régénérative et d’hyperleucocytose, mais, réticulocytes et polynucléaires activés sont particulièrement adhérents contribuant ainsi largement à la vaso-occlusion. Le traitement comportera une antibiothérapie associant une érythromycine (clarythromycine de préférence) à l’amoxicilline ou une céphalosporine, l’oxygénothérapie, une hydratation modérée de 2l/m2 et le plus souvent une petite transfusion de 10 ml/kg sera prescrite avant de discuter un éventuel échange si l’infiltrat et les signes cliniques progressent
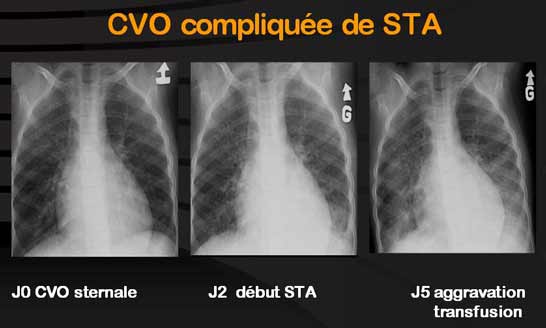
Les accentuations de l’anémie : il est indispensable de connaître pour chaque patient sa numération formule sanguine de base. Les drépanocytaires SS ou Sβ0 ont une Hb autour de 8g alors que les SC et Sβ+ sont plus proches de 10g. L’anémie jusqu’à un certain degré est bien supportée par le drépanocytaire à la différence du thalassémique qu’il faut maintenir > 9g pour une bonne croissance. Cependant des études récentes montrent que les drépanocytaires avec anémie intense chronique avec Hb<7g sont à haut risque de déficience cognitive et sont exposés à un plus grand nombre de risques (décès précoce, risque accru d’AVC). De façon aigue, l’anémie peut s’accentuer par hémolyse accrue en particulier au cours des CVO ou par crise arégénérative comme on l’observe surtout lors des infections à Parvovirus : le taux de réticulocytes des patients SS est autour de 250000 en moyenne et tout taux < 100000 doit faire évoquer le Parvovirus vis à vis duquel l’apparition d’IgG protège ensuite pour la vie. Cette crise arégénérative va être responsable d’une intensification de l’anémie nécessitant dans la grande majorité des cas une voire 2 transfusions d’autant que cette infection est souvent responsable de l’apparition d’une CVO voire d’un STA ou d’une séquestration splénique. Une vaccination serait très souhaitable mais non disponible à l’heure actuelle.
Le risque d’AVC clinique ne concerne heureusement que 10% environ des enfants SS, Sβ0 chez lesquels peuvent survenir convulsions, hémiparésies, paralysies crâniennes…de façon spontanée ou surtout au cours d’une CVO et encore plus au décours d’un STA. Le plus souvent, les signes neurologiques moteurs et sensitifs régressent mais des séquelles cognitives persistent et en absence de traitement adapté, le risque de récidive est majeur (de 67%) avec cette fois un risque de séquelles motrices majeur. Par contre, la mise en route d’un programme transfusionnel majeur visant à maintenir le taux d’HbS < 30% permet une réduction du risque de récidive à 10%. Dans la grande majorité des cas (75%), ces AVC sont de nature ischémique et en rapport avec une vasculopathie des artères cérébrales (artères cérébrales moyennes ACM et cérébrales antérieures ACA) détectable par Doppler transcrânien (DTC) : cet examen est devenu indispensable dans le suivi des enfants SS et Sβ0 et ceci dès l’âge de 1 an, puisque la vasculopathie peut apparaître dès cet âge.
Le risque de priapisme, concerne tous les garçons SS, SC et Sβthal ; il est heureusement assez rare dans l’enfance mais pourvoyeur d’un risque majeur fonctionnel ultérieur ; il se traduit par une érection anormale, parce que douloureuse et prolongée, nécessitant une prise en charge hospitalière rapide : les injections intracaverneuses d’Effortil utilisées depuis quelques années ont souvent permis la déturgescence et limité les indications d’échanges transfusionnels.
©F Bernaudin, S Verlhac - Mars 2006
Charger cette page au format pdf (371 kO), cliquez sur ce lien : Clinique drépanocytose
Adams R, McKie V, Nichols F et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N E J Med 1992, 326, 9, 605-610
Adams RJ, McKie VC, Hsu L et a l: Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 1998; 339: 5-11.
Bernaudin F, S.Verlhac, F. Fréard et al. A multicenter prospective study of children with sickle cell disease: radiographic and psychometric correlation PHRC 95. J Child Neurol 2000,15: 333-343
Galactéros F. Détection néonatale de la drépanocytose en France métropolitaine. Path Biol, 1999, 47, 1: 13-18
Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med 2000 Jan 13;342(2):83-9
Nagel RL. Severity, pathobiology, epistatic effects and genetic markers in sickle cell anemia. Seminars in hematology 1991, 28, 3, 180-201
Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, Wethers DL, Pegelow CH, Gill FM. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998 Jan 1;91(1):288-94
Platt OS, et al : Mortality In Sickle Cell Disease -- Life Expectancy and Risk Factors for Early Death. NEJM, 1994 330, 23
Powars DR. Sickle cell anemia : bs-gene-cluster haplotypes as prognostic indicators of vital organ failure. Seminars in hematology 1991, 28, 3, 202-208
Russell MO, Goldberg HI, Hodson A, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrencce of stroke in Sickle Cell Disease. Blood 1984; 63, 1: 162-169
Verlhac S, Bernaudin F, Tortrat D, et al: Detection of cerebrovascular disease in Sickle Cell Disease children by transcranial doppler sonography. Correlation with MRI and MRA and conventionnal angiography. Pediatric Radiology 1995, 25: S14-S19

